Leading the way in Research Lab Innovations
The Liotta Research Group
Discovering novel therapeutics for a healthier world.
Leading Research in Drug Discovery
Our Vision:
Our goal is to create novel medicines that transform human health. We aim to reinvent academic drug discovery, as the most formidable unmet medical needs don’t fit neatly into one bucket.
Innovative Drug Discovery Solutions
CXCR4 Antagonists
NMDAR Modulators
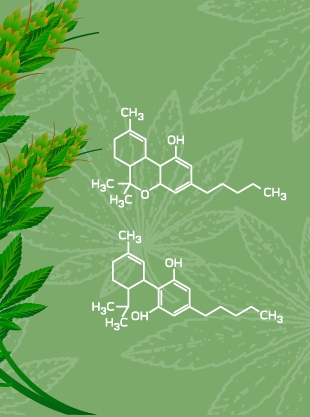
CBD/THC Prodrugs

Neurosteroid/Psychedelic Prodrugs
Lipid Prodrugs
Empower Innovation
See Dr. Liotta speak about his work.
Learn more about academic drug discovery research.
Dr. Liotta speaks at the 2024 UIC Drug Discovery Symposium
Two Pandemics and Two Drugs Developed to Control Them.
Hear the story of molnupiravir and how lessons from the HIV pandemic ultimately lead to its discovery.