
Authors: Jørn Hansen, Jochen Autschbach and Huw M. L. Davies
J. Org. Chem.,
2009, 74 (17), 6555–6563
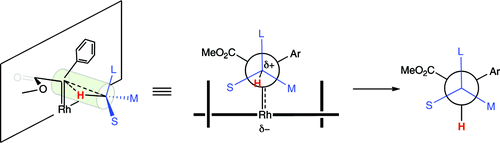
The mechanism of rhodium-catalyzed cyclopropanation and C−H functionalization reactions with methyl phenyldiazoacetate and methyl diazoacetate has been studied computationally with DFT. In accordance with experimental data, it has been demonstrated that donor/acceptor rhodium carbenoids display potential energy activation barriers consistent with the much higher selectivity in cyclopropanation and C−H insertion chemistry compared to the traditionally used acceptor carbenoids derived from unsubstituted diazo esters. Significantly higher potential energy barriers were found for transformations of donor/acceptor carbenoids than for those of acceptor systems, primarily due to the inherent stability of the former. Analyses of transition state geometries have led to the development of a rational model for the prediction of the stereochemical outcome of intermolecular C−H insertions with donor/acceptor rhodium carbenoids.