
Authors: Zhanjie Li, Vyacheslav Boyarskikh, Jørn H. Hansen, Jochen Autschbach, Djamaladdin G. Musaev , and Huw M. L. Davies
J. Am. Chem. Soc.,
2012, 134 (37), 15497–15504
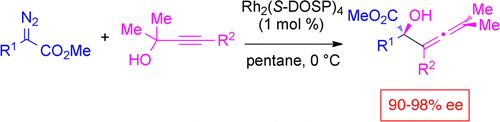
Rhodium-catalyzed reactions of tertiary propargylic alcohols with methyl aryl- and styryldiazoacetates result in tandem reactions, consisting of oxonium ylide formation followed by [2,3]-sigmatropic rearrangement. This process competes favorably with the standard O–H insertion reaction of carbenoids. The resulting allenes are produced with high enantioselectivity (88–98% ee) when the reaction is catalyzed by the dirhodium tetraprolinate complex, Rh2(S-DOSP)4. Kinetic resolution is possible when racemic tertiary propargylic alcohols are used as substrates. Under the kinetic resolution conditions, the allenes are formed with good diastereoselectivity and enantioselectivity (up to 6.1:1 dr, 88–93% ee), and the unreacted alcohols are enantioenriched to 65–95% ee. Computational studies reveal that the high asymmetric induction is obtained via an organized transition state involving a two-point attachment: ylide formation between the alcohol oxygen and the carbenoid and hydrogen bonding of the alcohol to a carboxylate ligand. The 2,3-sigmatropic rearrangement proceeds through initial cleavage of the O–H bond to generate an intermediate with close-lying open-shell singlet, triplet, and closed-shell singlet electronic states. This intermediate would have significant diradical character, which is consistent with the observation that the 2,3-sigmatropic rearrangement is favored with donor/acceptor carbenoids and more highly functionalized propargylic alcohols.