|
Photocatalysis at semiconductor-liquid interfaces represents a complex process, which includes band bending, built-in electric fields, surface recombination of photoexcited carriers, and ultimately charge transfer to the ions in solution, in which the electrostatic fields within the double layer plays an important role. There are loss mechanisms associated with each of these key components in the overall photoconversion efficiency. However, this complex process is often oversimplified in order to provide a basic interpretation of experimental data, largely because simple methods for separating these key components do not yet exist. In this collaborative project, Prof. Tim Lian’s and Steve Cronin’s groups will apply spectroscopic tools to study and separate the key components of the photocatalytic process, and to systematically vary precisely-defined photocatalytic structures in order to improve our understanding of energy loss in this important energy conversion system. Specifically, this proposal will explore both charge carrier dynamics and local electrostatic fields at photocatalytic interfaces using transient reflectance spectroscopy and Stark-shift surface enhanced Raman scattering spectroscopy, respectively. These techniques probe the charge carriers within the semiconductor and the electrostatic fields in the electrolyte double layer, both of which have been elusive experimentally. This proposed effort addresses the following questions regarding the charge carrier dynamics and electrostatic fields at these complex photoelectrochemical interfaces:
|
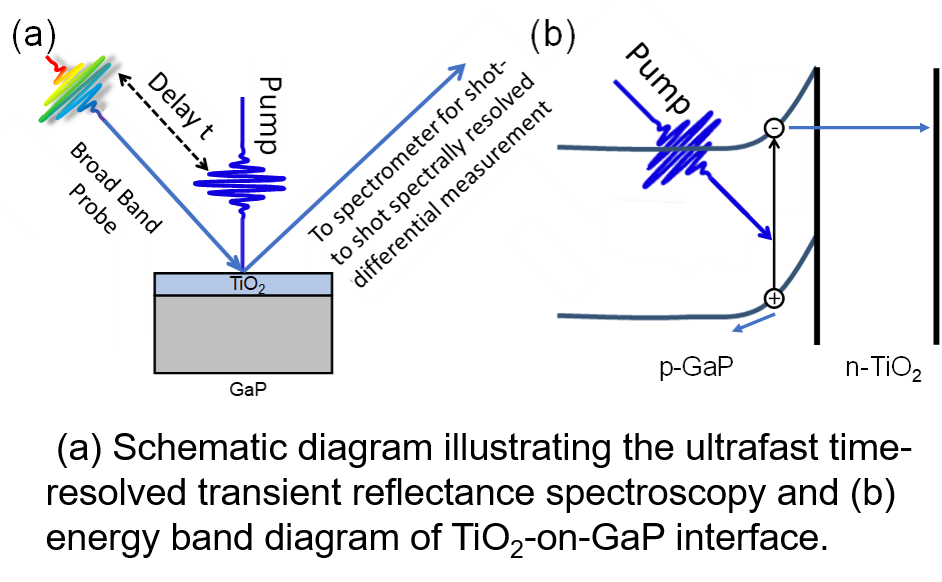

|
1) What is the extent to which the local electrostatic field at an electrode surface affects the potential required to drive reactions?
2) What is the role of the amorphous TiO2 layer in affecting the solid-state system and the chemical system (i.e., pn-junction formation vs. catalytically active sites)?
3) What is the fundamental nature of the rather slow kinetics (~µsec) observed in these photoelectrodes in solution?
If successful, the proposed work represents a substantial step towards realizing a rigorous and detailed understanding of the photocatalytic process and providing the insight needed to establish a rigorous theoretical understanding of the solid-state system and interfacial chemistry that has been lacking for decades. This fundamental understanding will, in turn, enable more efficient photocatalysts to be developed and engineered.
We acknowledge generous financial support by DOE
