Homeostatic Plasticity In Living Embryonic Networks
When spiking activity within a network is perturbed for hours to days, compensatory changes in synaptic strength or intrinsic cellular excitability are triggered that are thought to be important for the homeostatic maintenance of network or cellular spiking activity (homeostatic plasticity). We find that spontaneous network activity expressed in the embryonic spinal cord is homeostatically maintained following different perturbations. We have identified several different homeostatic mechanisms following the blockade of either spiking or GABAergic neurotransmission in the living embryo; we have determined that these perturbations trigger synaptic scaling of both glutamatergic and GABAergic synapses, as well as fast changes in intrinsic cellular excitability. We find that GABAergic scaling occurs through intracellular chloride regulation, that glutamatergic scaling occurs through insertion of calcium-permeable glutamate receptors, and that homeostatic changes in intrinsic excitability are mediated by changes in voltage gated channel currents (Na+, K+). We are continuing to characterize these plasticity mechanisms, but are also focusing on the triggers that initiate such plasticity. We are particularly focused on the possibility that reduced GABAergic transmission initiates the signaling pathways that underlie the homeostatic response. Understanding the compensatory plasticity that is triggered following perturbations to SNA (e.g. following fetal exposure to GABAergic modulators) will be important in understanding and avoiding disorders of the developing nervous system.
Optogenetic Multi-Electrode Approaches to
Homeostatic Plasticity
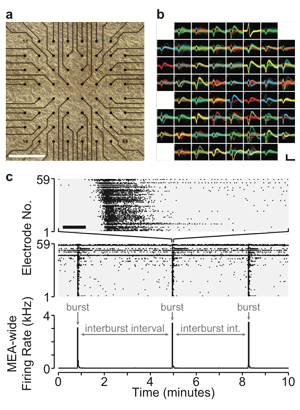
Homeostatic plasticity represents a set of mechanisms that ensures cells or networks maintain appropriate levels of spiking activity. The best-studied form of homeostatic plasticity is synaptic scaling, where quantal amplitudes throughout the cell are multiplicatively altered in a compensatory direction following chronic changes in spiking activity. It is generally believed that reductions in spiking trigger upward scaling, which then functions to homeostatically recover spiking levels. Instead, we have recently observed that AMPAergic upscaling appears to function to maintain synaptic strength, rather than spiking activity. The finding suggests other plasticities must homeostatically control spiking, and argues that synaptic strength itself may be homeostatically controlled at individual synapses. We have discovered this important finding by separately manipulating spiking and neurotransmission using a combination of multi-electrode array (MEA) recording, closed-loop optogenetic stimulation, and pharmacology of cultured cortical neurons. We can measure spiking activity in cortical cultured networks over days, as well as set target activity levels by driving activity optogenetically, based on feedback from the MEA recordings. Future studies will identify the importance of spiking and neurotransmission in other forms of scaling and homeostatic changes in intrinsic cellular excitability. Identifying the triggers and functional goals of these compensatory mechanisms will be crucial for understanding a network’s response to neural injury.
Mechanisms of Spontaneous Network Activity in the
Developing Spinal Cord
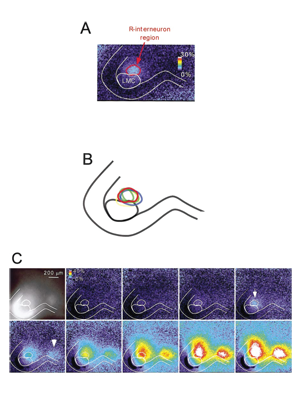
Spontaneous network activity (SNA) is experienced in virtually all circuits at some point during development (spinal cord, retina, hippocampus, brain stem, superior colliculus). SNA is expressed as episodic bursts of spiking activity that occur shortly after synaptic connections first form, at a time when GABA is excitatory. In the developing spinal cord embryonic limb movements are generated by SNA, which is known to be important in motoneuron axonal pathfinding, muscle development, and synaptic maturation. We are studying how SNA is regulated. We have identified several different mechanisms that are involved in the expression of this activity: presynaptic regulation of glutamatergic terminals by endocannabinoids, nicotinic regulation of SNA by presynaptic control of GABA release and through the activation a particular class of spinal interneuron. In addition, we find that intracellular chloride regulation, which alters the strength of GABAergic currents, is modulated by episodes of SNA and contributes to the excitability of the embryonic cord. We are now studying the transporters, which make GABAergic transmission more excitatory as they accumulate chloride during embryonic development and after spinal cord injury. By identifying the mechanisms that regulate SNA, we hope to better understand the maturation of spinal circuitry (excitability, synaptic strength, ionic gradients).