
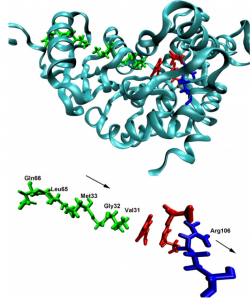
Proteins have highly dynamic structures, despite the beautiful appearance of static snapshots obtained from crystallographic models. We are investigating the role of protein motions in enzymatic catalysis. A question of intense current scrutiny is whether such motions are purely stochastic, simply serving to move the system towards conformational substates that are catalytically competent, or do protein motions couple more directly to the reaction coordinate? We focus on the dynamical nature of hydride transfer in model systems, lactate dehydrogenase (LDH) and dihydrofolate reductase (DHFR). We ask what types of atomic motion take place, how are the motions defined structurally, on what time scales do they occur, and how do these motions affect function?
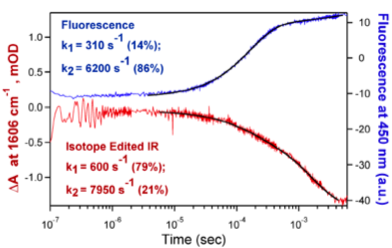
Our approach employs laser induced temperature jump and time-resolved, isotope-edited IR or time-resolved fluorescence spectroscopy to follow atomic motion from 10 ps to any slower time. For example, we have followed the fast relaxation kinetics of inner complex interconversion, LDH/NADH/pyruvate ↔ LDH/NAD+/lactate as shown in the figure to the right. We are also investigating mutations of the active site residues that change kcat and kcat/Km in known ways and in ways thought to be influenced by the protein dynamics. Relative motions between substrate and protein are characterized using isotope-edited time resolved IR absorption studies of specific substrate groups: for example, the C=O stretch of pyruvate and the antisymmetric stretch mode of the carboxylate group of pyruvate and lactate. Hydride and proton transfer kinetics are monitored using the changes in NADH emission near 450 nm as NAD+ converts to NADH, and the changes in pyruvate’s C=O stretch frequency and stretch modes of lactate as lactate converts to pyruvate. Kinetic isotope effects for the chemical processes provide further insight into the protein dynamics.
Funding
NIH P01 GM068036-011, “Protein Architecture and Energy Landscape of Catalysis,” (Callender, Dyer, Schramm, Schwartz co-PI’s);
6/2014-5/2019
Recent Publications
- “Acceleration of catalysis in dihydrofolate reductase by transient, site-specific photothermal excitation,” R. Kozlowski; J. Zhao; R.B. Dyer. PNAS, 2021, link
- “Site-Specific Tryptophan Labels Reveal Local Microsecond–Millisecond Motions of Dihydrofolate Reductase,” M.B. Vaughn; C. Biren; Q. Li; A. Ragupathi; R.B. Dyer. Molecules, 2020, link
- “Small molecule cores demonstrate non-competitive inhibition of lactate dehydrogenase,” B. Andrews, R.B. Dyer Med. Chem. Comm., 2018 Accepted link
- “Characterizing the Surface Coverage of Protein-Gold Nanoparticle Bioconjugates,” R. B. Kozlowski; A. Ragupathi; R.B. Dyer Bioconjugate Chemistry, 2018, 29, 2691–2700. link
- “Activity-Related Microsecond Dynamics Revealed by Temperature-Jump Förster Resonance Energy Transfer Measurements on Thermophilic Alcohol Dehydrogenase,”Vaughn, M. B.; Zhang, J.; Spiro, T. G.; Dyer, R. B.; Klinman, J. P., Activity-Related Microsecond Dynamics Revealed by T-Jump FRET Measurements on Thermophilic Alcohol Dehydrogenase. J Am Chem Soc 2018, 140, 900–903. link
- “Resolution of Submillisecond Kinetics of Multiple Reaction Pathways for Lactate Dehydrogenase”, M. J. Reddish, R. Callender, and R. B. Dyer, Biophys. J. 2017, 112(9), 1852-1862
- “Dual time-resolved temperature-jump fluorescence and infrared spectroscopy for the study of fast protein dynamics”, C. M. Davis, M. J. Reddish and R. B. Dyer, Spectrochim. Acta Mol. Biomol. Spectrosc. 2017, 178, 185-191
- “Ligand Dependent Conformational Dynamics of Dihydrofolate Reductase”, M. J. Reddish, M. B. Vaughn, R. Fu, and R. B. Dyer, Biochemistry. 2016, 55 (10), 1485–1493
- “The pathway of O2 to the active site in heme–copper oxidases”, Einarsdóttir, Ó., W. McDonald, C. Funatogawa, W.H. Woodruff, and R.B. Dyer, Biochim. Biophys. Acta – Bioenerg., 2015, 1847, 109-118.
- “The Dynamical Nature of Enzymatic Catalysis”, R. Callender and R. B. Dyer, Acc. Chem. Res., 2015, 48(2), 407-413.
- “Direct Evidence of Catalytic Heterogeneity in Lactate Dehydrogenase by Temperature Jump Infrared Spectroscopy”, M. J. Reddish, H.-L. Peng, K. S. Panwar, R. Callender, and R. B. Dyer, J. Phys. Chem. B, 2014, 118(37), 10854-10862.
- “Energy Landscape of the Michaelis Complex of Lactate Dehydrogenase: Relationship to Catalytic Mechanism”, H.-L. Peng, H. Deng, R.B. Dyer, and R. Callender, Biochemistry, 2014, 53(11), 1849-1857.
- “Anisotropic energy flow and allosteric ligand binding in albumin”, G. Li, D. Magana, and R.B. Dyer, Nature Communications, 2014, 5, 3100:1-7.