
Research in the Dyer lab concentrates on two aspects of solar energy conversion to chemical fuels: light induced multi-electron chemistry and interfacial charge transfer in nano-particle/enzyme hybrids. Although these are different projects, they have been coadjutant in their progress and design.
Driving Multi-Electron Chemistry with Light
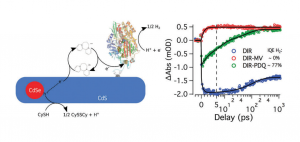
Nearly all carbon neutral chemical fuels (H2, O2, MeOH) require multi-electrons or holes for generation from stable precursors (H+, H2O, CO2). In order to drive these reactions with light electrons, or holes, must be delivered to catalytic species rapidly to eliminate reactive intermediate degradation. Our lab has developed photo-sensitization methods for driving enzymatic catalysis of hydrogenases (H2ases), enzymes that catalyzed the reduction of protons to hydrogen at rates as fast as 30,000 s-1. The use of electrostatic coupling to assemble negatively charged CdTe quantum dots to H2ase from Thiocapsa roseopersicina has shown photo-driven H2 production efficiencies of 4%. This hybrid system coupled with time resolved and steady state pump-probe experiments has allowed us to investigate the enzymatic mechanism in unprecedented detail by delivering electrons on the catalytically relevant timescale. We are currently extending these QD assemblies to other enzymes and catalysts of interest in renewable fuel generation and multi-electron chemistry.
Catalyst Photo-sensitizer Interfaces
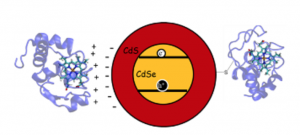
The development of efficient photo-sensitizer/catalyst hybrids has required rational design in the choice of material and the type of interface interaction being utilized. The objective has been to optimize the nanoparticle/metalloprotein interfacial interaction to promote the electron transfer with minimal intra-photo-sensitizer recombination. To achieve this, it is necessary to better understand the nano-particle/metalloprotein interface by investigation the various schemes for association and directed binding. As a result we exploit the electrostatic and covalent binding modes for biasing metalloprotein orientation and electronic coupling with core/shell quantum dots for ultrafast charge transfer. This work has allowed us to optimize previously established enzyme-nanoparticle assemblies by rational design and we are currently exploring new novel mechanisms of coupling enzymes as well as synthetic catalysts to nano-particle surfaces.
Funding
NSF DMR 1409851, (Dyer, PI) “Functional Hybrid Biotic/Abiotic Materials,” ![]() 8/2014 – 9/2017
8/2014 – 9/2017
Recent Publications
- “Time-Resolved Infrared Spectroscopy Reveals the pH-Independence of the First Electron Transfer Step in the [FeFe] Hydrogenase Catalytic Cycle” M. L. K. Sanchez; S. Wiley; E. Reijerse; W. Lubitz; J. A. Birrell; and R. B. Dyer. J. Phys. Chem. Lett. 2022, 13, 25, 5986–5990.
- “Efficient, Light-Driven Reduction of CO2 to CO by a Carbon Monoxide Dehydrogenase–CdSe/CdS Nanorod Photosystem” D. W. White; D. Esckilsen; S. K. Lee; S. W. Ragsdale; and R. B. Dyer. J. Phys. Chem. Lett. 2022, 13, 24, 5553–5556.
- “The Laser-Induced Potential Jump: A Method for Rapid Electron Injection into Oxidoreductase Enzymes,” M.L.K. Sanchez; S.E. Konecny; S.M. Narehood; E.J. Reijerse; W. Lubitz; J.A. Birrell; R.B. Dyer. J. Phys. Chem. B., 2020, 124, 40, 8750–8760.
- “Surface-Ligand “Liquid” to “Crystalline” Phase Transition Modulates the Solar H2 Production Quantum Efficiency of CdS Nanorod/Mediator/Hydrogenase Assemblies,” W. Yang; G.E. Vansuch; Y. Liu; T. Jin; Q. Liu; A. Ge; M.L.K. Sanchez; D.K. Haja; M.W.W. Adams; R.B. Dyer; T. Lian. ACS Applied Materials & Interfaces, 2020, 12, 31, 35614–35625.
- “Metal–ligand cooperativity in the soluble hydrogenase-1 from Pyrococcus furiosus,” G. E. Vansuch; C. Wu; D. K. Haja; S. A. Blair; B. Chica; M. K. Johnson; M.W.W. Adams; R.B. Dyer. Chemical Science, 2020, 11, 8572-8581.
- “An Abrupt Change of Configuration of Surface Ligands Affects the H 2 Production Efficiency of Mediator-Based CdS Nanorod/Hydrogenase Assemblies,” W. Yang; G.E. Vansuch; Y. Liu; T. Jin; A. Ge; M.L.K Sanchez; R.B. Dyer; T Lian. 236th ECS Meeting (October 13-17, 2019)
- “Investigating the Kinetic Competency of CrHydA1 [FeFe] Hydrogenase Intermediate States via Time-resolved Infrared Spectroscopy,” M.L.K. Sanchez; C. Sommer; E. Reijerse; J.A. Birrell; W. Lubitz; R.B. Dyer. JACS, 2019, 141, 40, 16064–16070.
- “Optimizing electron transfer from CdSe QDs to hydrogenase for photocatalytic H2 production,” M. Sanchez; Wu, C.; Adams, M.W.W.; R.B. Dyer. ChemComm, 2019, 55, 5579-5582.
- “Applications of Photogating and Time Resolved Spectroscopy to Mechanistic Studies of Hydrogenases,” Greene, B.L.; Vansuch, G.E.; Adams, M.W.W.; Dyer, R.B. Accounts of Chemical Research, 2017, 50, 2718–2726.
- “Balancing electron transfer rate and driving force for efficient photocatalytic hydrogen production in CdSe/CdS nanorod-[NiFe] hydrogenase assemblies,” Chica, B.; Wu C.; Liu, Y.; Adams, M.W.W.; Lian T.; Dyer, R. B. Energy & Environmental Science, 2017,10, 2245 – 2255.
- “Pre-Steady-State Kinetics of Catalytic Intermediates of an [FeFe]-Hydrogenase”, B. L. Greene, G. J. Schut, M. W. Adams and R. B. Dyer, ACS Catalysis 2017, 7(3), 2145-2150.
- “Glutamate Gated Proton-Coupled Electron Transfer Activity of a [NiFe]-Hydrogenase”, B. L. Greene, G. E. Vansuch, C. H. Wu, M. W. Adams and R. B. Dyer, J. Am. Chem. Soc. 2016, 138 (39), 13013–13021
- “Proton Inventory and Dynamics in the Nia-S to Nia-C Transition of a [NiFe] Hydrogenase”, B. L. Greene, C. H. Wu, G. E. Vansuch, M. W. Adams and R. B. Dyer, Biochemistry 2016, 55 (12), 1813–1825
- “Proton-Coupled Electron Transfer Dynamics in the Catalytic Mechanism of a [NiFe]-Hydrogenase.”, B. L. Greene, C.-H. Wu, P.M. McTernan, M.W.W. Adams, and R.B. Dyer, J. Am. Chem. Soc., 2015, Articles ASAP.
- “CO2 Reduction Catalyzed by Mercaptopteridine on Glassy Carbon.”, D. Xiang, D. Magana, and R.B. Dyer, J. Am. Chem. Soc., 2014, 136(40): p. 14007-14010
- “Direct evidence of active site reduction and photo-driven catalysis in sensitized hydrogenase assemblies,” R. Dyer, B. L. Greene C. A. Joseph M. J. Maroney J. Am. Chem. Soc. 2012, 134, 11108−11111. PMC3394927
- “Time-resolved vibrational spectroscopy detects protein-based intermediates in the photosynthetic oxygen-evolving cycle,” B. A. Barry, I. B. Cooper, A. DeRiso, S. H. Brewer, D. M. Vu, R. B. Dyer, Proc. Natl. Acad. Sci. USA 2006, 103, 7288-7291.