
By: Linda Huang, Shivani Seth, Shawn Kripalani, Anasua Bandyopadhyay and Nikita Maddineni
Imagine you’re living in the middle ages welding steel swords, leading your mundane life. Suddenly, your child is overcome with a horrid fever. Distraught, you also start to notice your child developing shockingly large lumps all over his body. What is going on? Soon, you learn that this nightmare is not only endangering your son’s life, but also the communities around you. Two weeks later, everyone around you has passed away, but for some reason you managed to survive. Why?
This so-called curse was actually known as the Black Plague that spread throughout Europe. The black death killed 75 to 200 million people and it peaked in Europe between the years of 1346 and 1353. It began with a rat infected with the bacterium Yersinia pestis. The bacteria infect humans by attacking the lymph nodes. They then begin to rapidly replicate, causing the lymph nodes to swell and become buboes. These buboes cause the immune system to go into septic shock, which is multiple-organ failure. This was one of the darkest points of European history, as 30-60% of Europe’s total population was killed. The methods of treatment ranged from visiting witch doctors to bathing in urine to turning to the church. Communities even started to live in the sewage systems after they became aware that it could be airborne. However, there was a small percentage of the population that was able to survive the plague. In the population that survived, researchers found that there was a mutation in the gene for the CCR5 cell membrane receptor called CCR5-∆32.
This is an example of natural selection, where the bubonic plague acted as the selection pressure for individuals with the mutation. These individuals with the delta-32 mutation had a higher fitness because they were able to survive and reproduce better compared to individuals who were susceptible to the black plague.
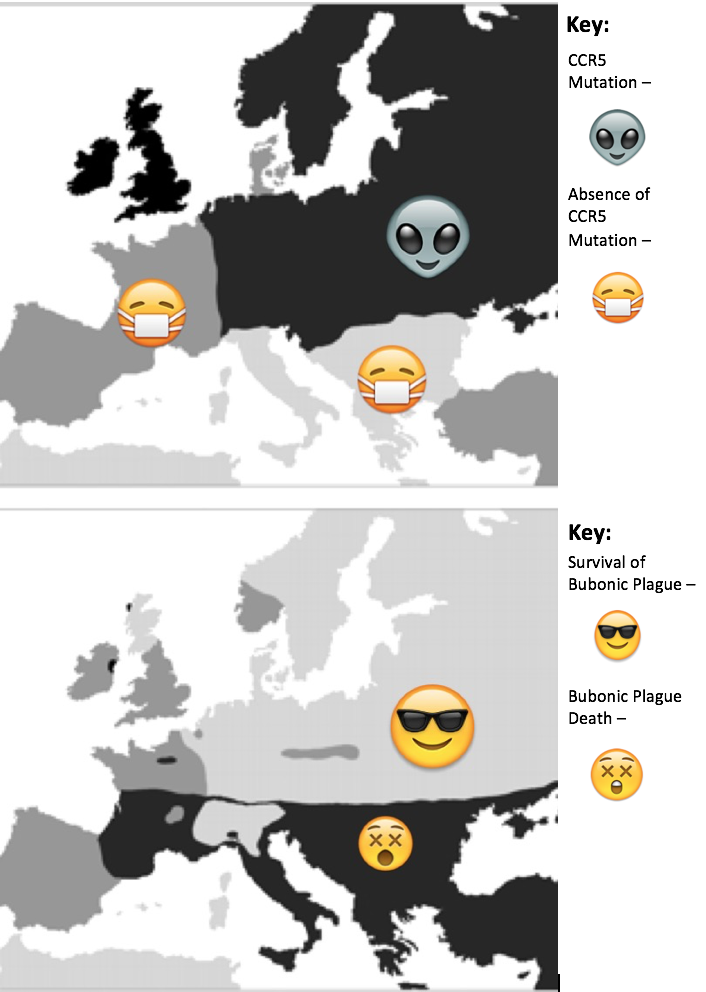
Figure 1: Top map shows regions of Europe during the black plague where the CCR5-∆32 mutation was present vs. regions where it was not present. Bottom map shows regions of Europe that were affected by the Black Plague vs regions that were not.
Coincidentally, research has shown that this mutation can also provide protection against HIV infection. In order for HIV to enter cells, the CCR5 receptor must be present on the surface of the cell. However, in individuals with the CCR5-∆32 mutation, the CCR5 receptor is not present on the cell membrane. Due to this mutation, individuals are resistant to HIV. This relationship between HIV and the CCR5-∆32 mutation is a way to debunk the misconception that all mutations are detrimental. In this case, having the mutation leads to increased fitness.
The protective effects of the CCR5 delta32 mutation against HIV infection are also being investigated as a potential long-term treatment option. Currently, antiretroviral therapy (ART) is the most widely used treatment for HIV, consisting of a combination of drugs that suppress progression of the disease and for reduce rates of transmission. However, ART is not a cure and individuals must rely on daily drug regimens for the entirety of their lives. Furthermore, the virus can eventually develop resistance to ART, leading to a need for more long-term treatment options.
In 2009, Hutter and other researchers identified an innovative approach using stem-cell transplantation. In a case study, a patient had HIV infection for ten years and acute myeloid leukemia. Researchers performed a stem-cell transplantation from bone marrow from an immune-compatible donor whose cells lacked expression of CCR5. The donor, who was homozygous for the CCR5 delta32 mutation, was resistant to the HIV infection. Researchers hoped that transplanting these cells with the mutation to the HIV-infected patient would confer resistance. After two transplantations, there was no recurrence of leukemia or detectable HIV in the bloodstream. The CD4+ T-cells in this patient have returned to the normal range and all carry the donor’s CCR5 gene. This patient has remained without any evidence of HIV infection for more than 8 years after discontinuation of ART, providing encouragement for stem cell transplantation as a more long-term treatment for HIV.
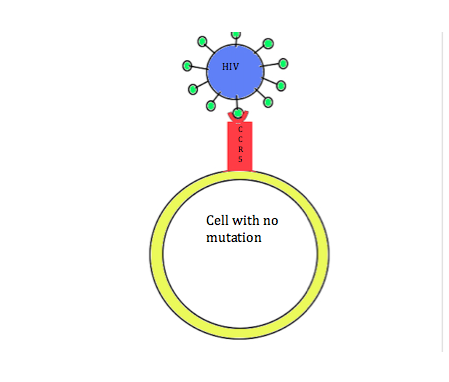
Figure 2A: Normal cell that has the CCR5 receptor. HIV can enter and infect the cell.
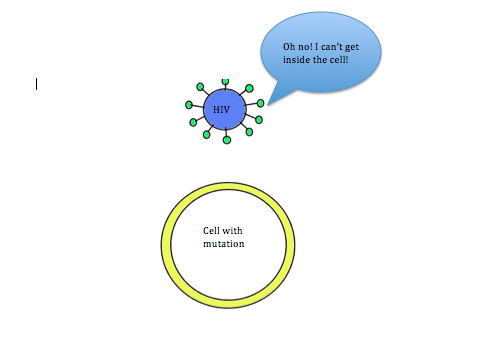
Figure 2B: Mutated cell without the CCR5 receptor. HIV is not able to enter and infect the cell, thus making the individual immune to the virus.
To learn more:
Petz, L.D., et al. 2015. Progress toward curing HIV infection with hematopoietic cell transplantation. Stem Cells Cloning 8: 109-116.
Cohn, S.K., & Weaver, L.T. 2006. The Black Death and AIDS: CCR5-Δ32 in genetics and history. Q J Med 99: 497-503.
Allers, K., et al. 2011. Evidence for the cure of HIV infection by CCR5Δ32/Δ32 stem cell transplantation. Blood 117(10): 2791-99.
Galvani, A.P., & Slatkin, M. 2003. Evaluating plague and smallpox as historical selective pressures for the CCR5-Δ32 HIV-resistance allele. PNAS 100(25): 15276-79.
Shariff, Mohammed. “10 Crazy Cures for the Black Death- Listverse.” List verse. N.p., 21 Jan. 2013. Web. 06 Dec. 2015.
Libert, F., et al. 1998. The CCR5 mutation conferring protection against HIV-1 in Caucasian populations has a single and recent origin in Northeastern Europe. Human Molecular Genetics 7(3): 399-406.
Stumpf, M.P., & Wilkinson-Herbots, H.M. 2004. Allelic histories: positive selection on a HIV-resistance allele. Trends in Ecology and Evolution 19(4): 166-8.
Hutter, G., et al. 2009. Long-Term Control of HIV by CCR5 Delta32/Delta32 Stem-Cell Transplantation. New England Journal of Medicine 360: 692-698.

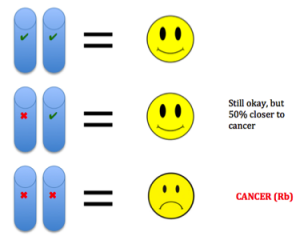 etic mutations and environmental exposures during one’s lifetime. Cancer is not heritable, but a predisposition to the disease can be (Peltomaki, 2012). For example, in our DNA we have two copies of each tumor suppressor gene, one from each parent. These genes keep cells from growing uncontrollably. So to lose function, one has to have mutations in both copies. Unfortunately, one can be born with a mutation in one copy, such as in Retinoblastoma (Rb). This means the gene is still expressed using the normal copy, but this confers a 50% increased predisposition for the disease bringing cells halfway closer to being cancer cells (Price et al., 2014).
etic mutations and environmental exposures during one’s lifetime. Cancer is not heritable, but a predisposition to the disease can be (Peltomaki, 2012). For example, in our DNA we have two copies of each tumor suppressor gene, one from each parent. These genes keep cells from growing uncontrollably. So to lose function, one has to have mutations in both copies. Unfortunately, one can be born with a mutation in one copy, such as in Retinoblastoma (Rb). This means the gene is still expressed using the normal copy, but this confers a 50% increased predisposition for the disease bringing cells halfway closer to being cancer cells (Price et al., 2014).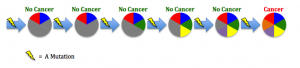 e different environments around cells fostering different mutations in various areas of the same tumor (Hardiman et al., 2015). Cancer is adapting to therapies that target mutations, making it so difficult to control.
e different environments around cells fostering different mutations in various areas of the same tumor (Hardiman et al., 2015). Cancer is adapting to therapies that target mutations, making it so difficult to control.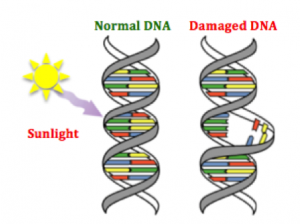 g agents are naturally occurring substances we encounter on a daily basis (Bauer, Corbett, & Doetsch, 2015). The human body has evolved ways to counteract DNA-damaging events, for example during sunlight exposure, using molecular machinery and likewise other naturally occurring compounds (Nishisgori, 2015).
g agents are naturally occurring substances we encounter on a daily basis (Bauer, Corbett, & Doetsch, 2015). The human body has evolved ways to counteract DNA-damaging events, for example during sunlight exposure, using molecular machinery and likewise other naturally occurring compounds (Nishisgori, 2015).




 can change genes, often leading to changes in the organism. For Oseltamivir resistance, a point mutation, the change of a single base of DNA, is all that is necessary. A change in the genes leads to a change in the amino acids produced. This change in the amino acids is just one amino acid at the 274th spot in the chain leads to a change in the activity of the virus. This line of reasoning is why the mutation is called H274Y.
can change genes, often leading to changes in the organism. For Oseltamivir resistance, a point mutation, the change of a single base of DNA, is all that is necessary. A change in the genes leads to a change in the amino acids produced. This change in the amino acids is just one amino acid at the 274th spot in the chain leads to a change in the activity of the virus. This line of reasoning is why the mutation is called H274Y.
